FDA Shifts Authorizations for Two COVID-19 Therapies Because of Omicron Subvariants
U.S. drug regulators have shifted language on two monoclonal antibody medicines because of emerging subvariants of the Omicron virus variant. The Food and Drug Administration (FDA) in recent days stipulated that an antibody treatment from GlaxoSmithKline and Vir Biotechnology cannot be used in regions where COVID-19 cases are likely to be caused by a variant “non-susceptible” to the drug. “Sotrovimab is not authorized for treatment of mild to moderate COVID-19 in geographic regions where infection is likely to have been caused by a non-susceptible SARS-CoV-2 variant, based on available information including variant susceptibility to this drug and regional variant frequency,” the FDA said in an updated emergency use authorization letter (pdf) to a GlaxoSmithKline executive. At this time, sotrovimab, the treatment, is still authorized in every U.S. region “until further notice,” the agency says. Several preprint studies published in February indicate that sotrovimab performs worse against BA.2, a subvariant of Omicron. Both are strains of SARS-CoV-2, which causes COVID-19 and is also known as the CCP (Chinese Communist Party) virus. Researchers with Columbia University tested samples from patients who were infected by the original CCP virus strain or had received a COVID-19 vaccine and found BA.2 exhibited “marked resistance to 17 of 19 neutralizing monoclonal antibodies tested, including S309 (sotrovimab),” which had retained a strong reaction against Omicron. Sotrovimab “was active against BA.2,” but showed decreased neutralizing ability, New York University researchers found. GlaxoSmithKline and Vir didn’t respond to requests for comment. The companies said on Feb. 10 that preclinical data suggested sotrovimab “retains neutralizing activity” against BA.2. The FDA appears to be preparing for the event that more studies show decreased resistance to the subvariant. A spokesman for the agency told The Epoch Times that the change gives the FDA “flexibility” to make sure sotrovimab is only administered in places where the treatment “is likely to provide benefit” to recipients. The shift also aligns the authorization for sotrovimab with updated language for the other monoclonals the FDA has authorized. The FDA restricted use of monoclonals from Regeneron and Eli Lilly in January based on emerging data that indicated the treatments didn’t perform well against Omicron. Several weeks later, the agency cleared a separate drug from Eli Lilly because that monoclonal appeared to work well against Omicron and BA.2. According to a federally-run genomic surveillance program, BA.2 was responsible for 1 percent of cases in the United States during the week ending on Feb. 5. Another Omicron subvariant, B.1.1.529, was pegged as causing about 30 percent of cases in the country. The FDA on Feb. 25, meanwhile, authorized a higher first dose of Evusheld, an antibody combination drug cleared for prevention of SARS-CoV-2, because of data that suggests the drug “may be less active against” Omicron subvariants. The authorized regimen was 150 milligrams of each of the two antibodies, tixagevimab and cilgavimab. It’s now 300 milligrams each. Patients who received the previously authorized dosage should get an additional dose of 150 milligrams of each of the antibodies as soon as possible to ensure maximum protection, according to the FDA. Evusheld, made by AstraZeneca, is authorized to prevent CCP virus infection in people with compromised immune systems. AstraZeneca didn’t respond to a request for comment.
U.S. drug regulators have shifted language on two monoclonal antibody medicines because of emerging subvariants of the Omicron virus variant.
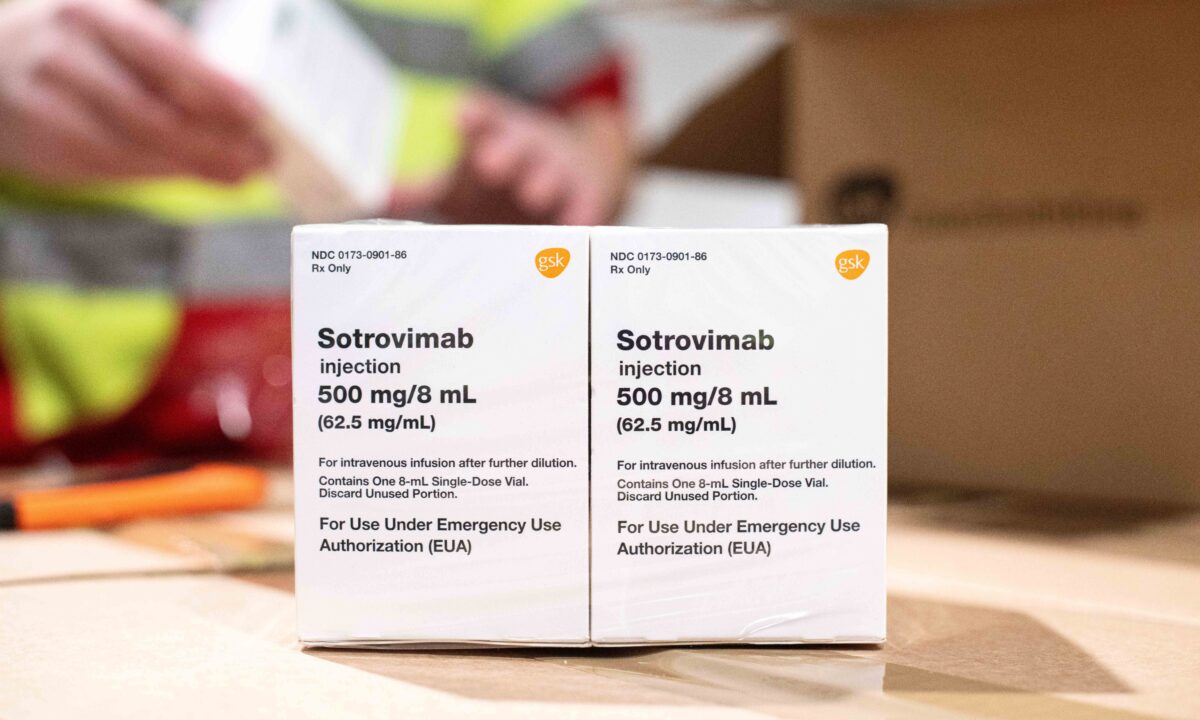
The Food and Drug Administration (FDA) in recent days stipulated that an antibody treatment from GlaxoSmithKline and Vir Biotechnology cannot be used in regions where COVID-19 cases are likely to be caused by a variant “non-susceptible” to the drug.
“Sotrovimab is not authorized for treatment of mild to moderate COVID-19 in geographic regions where infection is likely to have been caused by a non-susceptible SARS-CoV-2 variant, based on available information including variant susceptibility to this drug and regional variant frequency,” the FDA said in an updated emergency use authorization letter (pdf) to a GlaxoSmithKline executive.
At this time, sotrovimab, the treatment, is still authorized in every U.S. region “until further notice,” the agency says.
Several preprint studies published in February indicate that sotrovimab performs worse against BA.2, a subvariant of Omicron. Both are strains of SARS-CoV-2, which causes COVID-19 and is also known as the CCP (Chinese Communist Party) virus.
Researchers with Columbia University tested samples from patients who were infected by the original CCP virus strain or had received a COVID-19 vaccine and found BA.2 exhibited “marked resistance to 17 of 19 neutralizing monoclonal antibodies tested, including S309 (sotrovimab),” which had retained a strong reaction against Omicron. Sotrovimab “was active against BA.2,” but showed decreased neutralizing ability, New York University researchers found.
GlaxoSmithKline and Vir didn’t respond to requests for comment.
The companies said on Feb. 10 that preclinical data suggested sotrovimab “retains neutralizing activity” against BA.2.
The FDA appears to be preparing for the event that more studies show decreased resistance to the subvariant.
A spokesman for the agency told The Epoch Times that the change gives the FDA “flexibility” to make sure sotrovimab is only administered in places where the treatment “is likely to provide benefit” to recipients.
The shift also aligns the authorization for sotrovimab with updated language for the other monoclonals the FDA has authorized.
The FDA restricted use of monoclonals from Regeneron and Eli Lilly in January based on emerging data that indicated the treatments didn’t perform well against Omicron. Several weeks later, the agency cleared a separate drug from Eli Lilly because that monoclonal appeared to work well against Omicron and BA.2.
According to a federally-run genomic surveillance program, BA.2 was responsible for 1 percent of cases in the United States during the week ending on Feb. 5. Another Omicron subvariant, B.1.1.529, was pegged as causing about 30 percent of cases in the country.
The FDA on Feb. 25, meanwhile, authorized a higher first dose of Evusheld, an antibody combination drug cleared for prevention of SARS-CoV-2, because of data that suggests the drug “may be less active against” Omicron subvariants.
The authorized regimen was 150 milligrams of each of the two antibodies, tixagevimab and cilgavimab. It’s now 300 milligrams each.
Patients who received the previously authorized dosage should get an additional dose of 150 milligrams of each of the antibodies as soon as possible to ensure maximum protection, according to the FDA.
Evusheld, made by AstraZeneca, is authorized to prevent CCP virus infection in people with compromised immune systems.
AstraZeneca didn’t respond to a request for comment.